The model used by the TANGO algorithm is designed to predict cross-beta aggregation in peptides and denatured proteins and consists of a phase-space encompassing the random coil and 4 possible structural states: beta-turn, alpha-helix, beta-sheet aggregation and alpha-helical aggregation. Every segment of a peptide can populate each of these states according to a Boltzmann distribution, i.e. the frequency of population of each structural state for a given segment will be relative to its energy. Therefore, to predict cross-beta aggregating segments of a peptide TANGO simply calculates the partition function of the phase-space. Here we first describe how we determine the propensity for each of the different structural states, how we sample phase-space and which assumptions are embedded in these choices. Next we discuss how TANGO performs on a set of 176 peptides derived from 21 proteins.
Alpa-Helical propensities
The parameters used in the latest version of AGADIR (AGADIR-1s11), have been used to determine the helical propensity of the amino acid sequences. The only modification has been the implementation of a two window approximation (see below).
beta-Turn propensities
beta-turn propensity is calculated by considering four energy contributions: (1) an amino-acid specific cost in conformational entropy for fixing that residue in a beta-turn compatible conformation, (2) interactions of each amino acid with the turn structure in a position dependent manner, (3) in some cases side chain-side chain, or side chain-main chain interactions within the turn and (4) a single H-bond between the main chains of residues i and i+3 of the turn. We have only considered 4 types of turns for which we could obtain significant statistical data, Types I, I, II and II. The entropic cost of fixing a particular amino-acid in turn dihedral angles, has been obtained using statistical f,y matrices, as previously published. Since residues i and i+3 could adopt different conformations and are not fixed in the turn, we have applied a general entropy penalty term of 0.3 Kcal/mol at 298K. The interaction of the amino acids with the turn has been obtained by statistical analysis of the protein database (see methods section), assuming that counts for observed interactions higher than the expected value represent favorable interactions and the opposite is true.
Cross beta-aggregation
To estimate the aggregation tendency of a particular amino acid sequence, we have taken the following assumptions: (1) In an ordered beta-sheet aggregate the main secondary structure is beta-strand. (2) The regions involved in the aggregation process are fully buried, thus paying full solvation costs and gains, full entropy and optimize their H-bond potential (that is the number of H-bonds made in the aggregate is related to the number of donor groups that are compensated by acceptors). An excess of donors or acceptors remains unsatisfied. (3) Complementary charges in the selected window establish favorable electrostatic interactions and overall net charge of the peptide and net charges near the aggregating region (two residues before or after the choosen window), disfavor aggregation.
Estimation of beta-propensity.
We have included three energy contributions: a residue-specific cost in conformational entropy for fixing that residue in a beta-strand conformation and side chain-side chain interactions of residue i with residues at positions i+1 and i+2.
Formation of a beta-strand requires, in general, less conformational entropy cost than formation of an alpha-helix of equivalent length, because the beta-strand region of the Ramachandran plot is larger than the alpha-helical region while the depth of the energy well is similar. On the other hand, a single beta-strand does not have main chain-main chain hydrogen bonds that counteract the loss in conformational entropy. In the absence of other contributions the beta-strand will not be populated over the random coil. However, a factor not generally considered is the existence of intra-strand side chain-side chain interactions that when favorable could promote beta-strand population. The unique side chains that are close in space in an extended conformation (beta-strand) are those between positions i and i+2. Residues at positions i and i+1 could also influence the formation of the beta-strand since they are on average more distant than in the random-coil. This phenomenon has energetic implications that we denominate (i,i+1) beta-interactions. On this basis, favorable (i,i+1) b-interactions reflect repulsions between residues i and i+1 while (i,i+1) beta-unfavorable interactions reflect attractions of the these side chains when they are not in a beta-strand conformation. These side chain-side chain interactions introduce energetic coupling in the b-strand-coil transition, producing some cooperativity.
The entropic cost of fixing a particular aminoacid in beta dihedral angles, has been obtained using statistical f,y matrices, as previously published.
The other two terms participating in the equation, interaction between residues i,i+1 and i+2, are relative to the energy contribution of side chain-side chain interactions. They have been determined using a mean-force potential.
Desolvation costs of aggregated segments.
As explained above we assume that the residues forming the core of the ordered aggregate must be fully buried. This implies full desolvation and minimum degrees of freedom. The energetic cost of burying a sequence stretch is defined by the following equation:
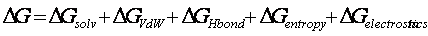
were Dsolv and Dvdw are obtained from the FOLD-EF forcefield (Reference) assuming maximum burial. DHbond is equal to the number of H-bonds made by the buried segment multiplied by the H-bond contribution (the same value used in AGADIR1s). The number of H-bonds is equal to the number of donors, or acceptors, in the polypeptide chain that could pair with an acceptor or donor, respectively. For the backbone this is always 2 per residue, and for the side chains we just count the total number of donors and acceptors and we take the minimum number of the two. In the case of Pro we consider that if it is N-terminal to the segment we loss only one backbone H-bond, while if it is C-terminal we loss two. A Pro inside a segment is penalized by 10 Kcal/mol.
Dentropy assumes full entropy cost and is the sum of the main chain entropy due to the residues being in an extended conformation and side chain entropy (as described by ABGYAN). The model used to calculate the electrostatic contribution to helix stability was previously described in Viguera, Lacroix, Serrano). In the following paragraph we describe how electrostatic contributions Delectrostatic to beta-aggregates are computed.
Electrostatic contribution.
The electrostatic interactions obviously change with the degree of ionization and consequently with the pH of the solution, while the pKa of ionizable groups in a peptide change from their standard values depending on the electrostatic environment. In TANGO we considered all electrostatic interactions (this involves charged side chain groups, free N-terminal and C-terminal main chain groups, and the succinyl blocking group if the peptide is succinylated) to compute the electrostatic environment of the amino acids in the random coil and in helical segments, taking into account the ionic strength, temperature and the pKa (see below).
TANGO distinguishes between charges in the segment under consideration (internal charges) which are considered fully buried, charges within two residues outside the N-or C-terminus of the segment (neighbouring charges) which are considered solvent exposed and the rest of the charges in the polypeptide chain (external charges). External charges are also considered to be solvent exposed but in addition their contribution is corrected with chain length. For buried charges we use a dielectric constant of (332/(8.8 * exp(-0.004314 * (temp-273.0)))), while for exposed charges it is 332/(88 * exp(-0.004314 * (temp-273.0))).
The net charge for the segment under consideration plus its neighbouring residues is calculated assuming an average distance between charges in the aggregate of around 5A. For the rest of the polypeptide chain TANGO calculates the net charge and divide it by the number of residues introducing a higher average distance for longer polypeptide chains.
There are two types of electrostatic interactions: repulsive interactions due to a net charge and attractive interactions due to compensated charges. The latter one has been introduced to reflect that on average some of the compensated charges will make salt bridges and thus contribute to the stability of the aggregate. In the case of the attractive compensated charges we correct the favorable electrostatic interaction calculated by dividing it by 3. This arbitrary correction factor is introduced since as explained above this term reflects the formation of internal salt bridges which of course cannot be formed by all compensated charges.
alpha-Helix aggregation.
Some peptides and proteins aggregate in a helical conformation. This is typically observed in proteins with a tendency to form coiled-coil structures or Leu-zippers (references). Since formation of dimers or higher order helical aggregates will compete with beta-sheet aggregation we have included this structural state in the TANGO algorithm in a very simple manner. As for beta-sheet aggregation we assume full burial upon aggregation, but only for one face of the helical structure. Thus, we assume than in a helical aggregate residues i,i+1, i+4,i+5, i+8, i+9 etc will be fully buried. For those residues we applied the same considerations as for burial of residues in beta-sheet aggregates. The energy required to fold the segment into a helical conformation, however, is directly derived from AGADIR.
The effect of physico-chemical conditions on aggregation
pH, ionic and temperature dependence
The effect of pH, temperature and ionic strength on electrostatic interactions was taken into account as described in AGADIR2-1s11. Similarly the dependence of entropy, H-bonds and hydrophobic interactions on temperature and ionic strength are taken into consideration as described in AGADIR2-1s11.
TFE dependence
The effect of TFE on the stability of the different structural conformations considered here has been taken into consideration in the following way. First, we assume a general increase of the H-bond contribution to the energies of the helical, turn and aggregated conformations (SEE REFERENCES IN BLANCO & SERRANO, PROT G). Second, we consider a change in the helical propensities of the amino acids based on the experimental results of Baldwin and co-workers. We assume that the effect of TFE is linear with concentration up to 40% were no further changes are considered. This is based on the empirical observation that for many peptides analyzed experimentally above 40% there are few changes if any.
Conformational sampling by a two-windows approximation.
Ideally to calculate a partition function a multiple window approximation as used for the AGADIRms algorithm and described in Munoz et al. should have been implemented. However, since we are taking into consideration 4 possible structural states, the calculation of the partition function would be computationally too demanding. Therefore we have opted for a two-window approximation which assumes that the probability of finding more than two ordered segments in the same polypeptide chain is too low to be considered (the simple one window will deviate too much from reality for peptides with > 50 residues). Our assumption is that in the same polypeptide chain there could be one or two non-overlapping (separated by 5 unstructured residues or more, see Munoz & Serrano, Biopolymers, AGADIR2-1s) structured segments. This simplification could therefore result in deviations for large proteins containing several strongly predicted structured regions.
A second simplification is that we do not consider aggregation intermediates. We consider aggregates as a single molecular species or structural state in competition with b-turn and a-helical conformations again for the sake of simplifying the partition function. This simplification can be translated in the assumption that the aggregating segment has an infinite concentration, or in other words, that once formed it immediately aggregates with infinite association constant. Since in reality the aggregation kinetics and the extent of aggregation will depend on the concentration of the peptide as well as of its association constant, this means that the aggregation probabilities we are obtaining are only relative. Thus they allow comparison inside the same polypeptide chain, or with mutants of the polypeptide chain, but not between different polypeptide chains.
Third, like in the multiple window approximation of AGADIR we have assumed that there is no energetic coupling between the two non-overlapping segments (independent of their conformation) that are simultaneously present in the same molecule. This assumption seems rather reasonable for monomeric peptides in which there are no long or medium range interactions. Finally, we assume that all possible states can coexist by pairs in the same polypeptide molecule, that is an aggregate can have a helical segment as long as it is out of the aggregated segment (there is experimental evidence for this, like in lysozyme were helical regions still persist in the amyloid aggregate; DOBSON).
Under these assumptions and the definition of the random coil state as those conformations which are not helical, or turn or involved in aggregation, the two-window sequence partition function becomes the sum of the statistical weights for all the possible combinations of structured segments (from one to two non-overlapping segments) plus the statistical weight for the random coil state (the set of molecular conformations which do not include any structured segment). The weight for the random coil is 1 (arises from the product of the weights of all the residues in the random coil state). As a result of the third assumption, the statistical weight of molecular conformations with more than one structured segment are simply the product of the weights of all the structured segments included on it (see Munoz et al.).


